
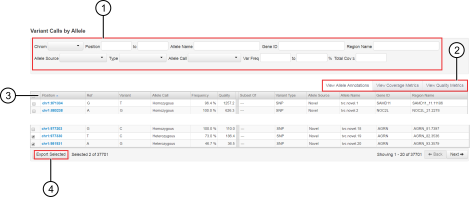
Variant Calls by Allele table
The detailed variantCaller plugin report contains the Variant Calls by Allele table. The table lists the details about each variant that is called, including the allele locus, allele annotation, coverage metrics, and quality metrics for the specific barcode or sample. You can use the table to find the variant alleles of interest and information about those alleles. You can also export the information to be saved to your local storage.
To access the report, click the sample name link in the Sample column in a report from a sequencing run.
-
Find the variants of interest by applying filters to the table to narrow down the list of variants called.
-
Change the display of the table to view allele annotation, coverage metrics, or quality metrics for each variant. To switch between different displays, you can select one of the following tabs.
-
View Allele Annotation. For more information, see View allele annotations .
-
View Coverage Metrics. For more information, see View coverage metrics .
-
View Quality Metrics. For more information, see View quality metrics.
-
-
Click the column heading to sort variant alleles by the values in the column.
-
Export the information that is associated with the selected variant alleles to an XLS file. The exported XLS file contains all the information about the selected variants, including the information listed in the View Allele Annotation, View Coverage Metrics, and View Quality Metrics tabs. The tabs that are available depend on the run type. For information about how to export the information, see Export variant calls to a file.
|
Column |
Description |
|---|---|
|
The chromosome (or contig) name in the reference genome, and the position of the chromosome (or contig) in the one-based coordinate. |
|
|
The reference base or bases. |
|
|
The variant allele base or bases. |
|
|
The zygosity (homozygouse or heterozygous) or type (absent or No Call) of the allele that is called by the zygosity (homozygouse or heterozygous) or type (Absent or No Call) of the allele all by the variantCaller plugin. |
|
|
The frequency, in %, of the variant allele. |
|
|
The limit of detection (LOD) at the genome location, estimated based on the number of detected molecules. This column is available only for sequencing runs that use the tag sequencing or Ion AmpliSeq™ HD as the target technique in the Planned Run. For more information, see Research application step in the workflow bar. |
|
|
The Phred-scored quality. For variants found by the Long INDEL Assembler, this value is always set to 50. Larger values mean higher confidence in the call. Quality is calculated by posterior probability that the variant allele frequency is greater than the cutoff (min_allele_freq in the parameter file), if a variant call is made, or posterior probability that the variant allele frequency is below this cutoff (if a reference call). The posterior probability that is computed as conditional on the reads observed includes sampling variability. Quality score is typically very large for reads strongly distinguishing variants with good depth, that is, under the model assumed, evidence is overwhelming for the variant or for the reference. Marginal values can mean that either the reads do not distinguish the variant well, there is insufficient depth to resolve, or the observed allele frequency is near the cutoff. |
|
|
An indication (0 or 1) of whether the variant allele is a possible polypoidy allele (PPA). Only absent alleles can be labeled as PPA; heterozygous and homozygous alleles are not treated as PPA. This column is available if the report_ppa parameter is set to 1. For more information on how to set the report_ppa parameter, see Torrent Variant Caller module advanced settings. |